BELLE Newsletter Vol. 5, No. 2, 1996
BELLE Debate
Is There a Need for a New Cancer Risk Assessment Paradigm?
Since the mid 1970's the field of cancer risk assessment has been dominated by the belief that both chemical and radiation induced cancer acts via a non-threshold mechanism. This belief has resulted in the acceptance and application of low dose linearity modeling methodologies to estimate risk in humans exposed to agents typically shown to be carcinogenic in animal bioassays.
Over the past 20 years the cancer risk assessment methodology has often been challenged but usually on a case by case approach. For example, the carcinogenicity of chloroform and saccharin in animal models is believed to be dose-tissue damage mediated responses which are not relevant to low dose linear modeling. Renal tumors following exposure to uncombusted gasoline is now recognized as a strain-specific, gender-specific response lacking in both generalizability to other species as well as to low dose linear modeling.
Other challenges to the current interpretation of cancer risk assessment estimates are more general with broader implications. For example, the substantial body of toxicological literature indicating that modest dietary restriction can profoundly reduce the occurrence of chemically induced tumors is forcing regulatory authorities to not only rethink the nature of the animal bioassay but also the risk assessment relevance of past studies.
An even broader challenge to the current risk assessment predictive scheme is emerging from the radiation literature suggesting that not only may low doses of radiation not be as harmful as once thought but that low doses may actually confer beneficial effects including increased longevity.
These various, diverse and complementary challenges to the current cancer risk assessment paradigm may have been dismissed in the past as being the product of ideologic bias, economic self-interest, or simply lacking in adequate data support. However, over the past decade each of the above challenges has become strengthened by a progressive series of mechanism-based findings which provide enhanced credibility for these perspectives.
Given the present developments in the field of cancer risk assessment it may be time to call the question "Is there a need for a new cancer risk assessment paradigm? And what should it be?
BELLE has asked a number of distinguished scholars to offer their thoughts on this topic. Those invited are intended to represent a broad spectrum of thought based on scientific interests and professional experiences.
Cancer and Non-Cancer Risk Assessment Should be Harmonized
Kenny S. Crump, Harvey J. Clewell
K.S. Crump Group, ICF Kaiser, 602 East Georgia Avenue,
Ruston, LA 71270; Tel; (318) 255-4800; Fax: (318) 255-4960
Melvin E. Andersen
ICF Kaiser, P.O. Box 14348, RTP, NC 27709;
Tel: (919) 547-1723; Fax: (919) 547-1710
ABSTRACT
Although fundamentally different risk assessment procedures have traditionally been applied to carcinogens and non-carcinogens, this dual approach does not have strong scientific support. We believe that this dichotomy between cancer and non-cancer risk policies has led to severe imbalances in the cost of regulation and the level of protection afforded by various regulations. Moreover, the practice of providing quantitative estimates of human risk from animal data has blurred the distinction between what is known and what is not known concerning risk to human populations, which has in turn caused misunderstanding and misuse of risk assessment by regulators and the general public. Therefore, we advocate harmonization of risk assessment practices for cancer and non-cancer endpoints by use of a common default risk assessment procedure that does not routinely involve making quantitative estimates of low-dose human risks from animal data.
PAST AND CURRENT PRACTICES
During the past 20 years risks from exposure to carcinogens and non-carcinogens have been assessed using fundamentally different approaches. Quantitative estimates of carcinogenic risks have been obtained by extrapolation to low doses using a linear dose-response model. Acceptable exposures to non-carcinogens have been estimated by applying uncertainty factors to NOAELs (no-observed-adverse effect levels). The practical outcome of this dual approach has been that chemicals determined to be carcinogenic, regardless of their mechanisms of action, have been regulated much more stringently than chemicals causing other types of toxicity.
Initially, the reason for using a linear dose-response with carcinogens was the belief that many carcinogens are mutagenic and therefore likely to act through direct interaction with DNA to produce a heritable change in a single cell that eventually produces a tumor. It seemed likely that the dose response for such a mechanism would not show a threshold but be linear at low doses (NRC, 1977). However, it now appears that many animal carcinogens may act indirectly through processes (e.g., cell killing) that are also involved in other forms of toxicity (Reitz et al. 1990; Beck et al. 1993; Larson et al. 1994). For these carcinogens the shape of the dose-response curve for cancer is apt to be similar to the dose-response curve for the underlying toxic response. If this toxic response is non-linear, the default use of a linear dose-response may cause a chemical to be regulated much more stringently than is commensurate with its toxicity.
It had been hoped that better understanding of carcinogenic mechanisms, coupled with mathematical models of cancer that incorporate mechanistic information, would provide better quantitative estimates of low-dose risks. However, our experience with such models indicates that this hope is unlikely to be realized any time soon. Perhaps the most revealing example of such modelling is EPA's biologically based cancer model for dioxin (EPA 1994a). The biologically-based model developed by EPA predicts essentially the same risk as the linearized multistage model used previously by EPA. That in itself is not discouraging. What is discouraging is the reason the two models produced the same answer. It is because a linear dose-response was assumed in both models. However, the underlying data are consistent with a linear, non-linear (Andersen et al., 1995) and even a threshold dose-response. Thus, even when using a biologically-based model, an assumption must be made regarding the shape of the dose-response at low doses. Much of the mechanistic information presently available informs the low dose extrapolation only in broad terms (e.g., by suggesting a linear or non-linear mechanism).
Although a threshold is generally assumed for non-cancer effects, the existence of a threshold cannot be proved. It has been argued that if a chemical acts by augmenting a process that is already producing cancer in unexposed subjects, then the dose-response is likely to be linear at low doses (Crump et al. 1976). Although this general argument has often been used to justify a linear dose-response for cancer, it applies equally well to non-cancer. For both cancer and non-cancer the shape of the dose-response at very low doses remains unknown and, perhaps, unknowable.
Differences in risk approaches for carcinogens and non-carcinogens also exist for steps other than low dose risk estimation. In animal-to-human extrapolation of non-cancer responses from oral dosing, animal doses are frequently converted to equivalent human doses on a body weight basis (mg/kg/day), and an additional uncertainty factor of 10 is applied. For carcinogens, on the other hand, this same conversion has been performed by assuming that equal exposures in units of mg/m2/day provides equal risks in animals and humans, although the argument used to support this approach (EPA, 1992) is just as applicable to non-cancer as to cancer. Another discrepancy between cancer and non-cancer approaches is the way in which human interindividual variability is handled. In non-cancer risk assessment, an uncertainty factor of 10 is often applied to protect sensitive individuals or subpopulations, and exposure guidelines have in some cases been based on data for a specific sensitive subpopulation, such as asthmatics. In contrast, the cancer risk assessment paradigm does not explicitly consider interindividual variability.
The dual approach to risk assessment assumes that a chemical can be classified unambiguously as a carcinogen or non-carcinogen. However, a recent analysis of the NTP cancer bioassay database (Crump et al. 1996) suggests that there are significantly more carcinogens in the database than were identified by the NTP. These undetected carcinogens caused increases in tumor response that were not sufficiently strong to be identified using conventional statistical techniques. There is no firm basis for believing that the risks to humans, if any, posed by these undetected carcinogens differ systematically from the risks, if any, posed by chemicals that caused a barely detectable increase in carcinogenic response. Yet, under the current procedures the undetected carcinogens are regulated far less stringently.
The emphasis upon quantitative estimates of cancer risk appears to have conveyed an overly optimistic sense of our knowledge concerning human risks from low exposures. For example, one of the provisions of the Clean Air Act Re-authorization uses a risk of 1/1,000,000 as a decision point. Although this provision implies that such risks can be estimated with at least some reasonable accuracy, this is not the case. Since estimates of the human exposure corresponding to a risk of 1/1,000,000 is determined in very large measure by the assumptions employed by the risk assessor, this requirement in the Clean Air Act results in policy being set by risk assessors in the guise of science. A number of acts proposed by the current Congress that would mandate extensive risk assessment also seem to be predicated upon an overly optimistic expectation of our ability to accurately quantify human risk.
The new EPA (1994b) draft guidelines for cancer risk assessment call for making a point estimate of the ED10 (dose corresponding to an additional cancer risk of 1/10). The ultimate default in the absence of contrary data is to estimate low-dose risks using a straight line from the ED10 through zero. However, if the carcinogenic mechanism is believed to be non-linear, a margin of exposure below the ED10 can be used to define an acceptable human exposure. Although these guidelines would make cancer and non cancer risk assessment appear more similar, important differences would still exist.
THE CASE FOR HARMONIZATION
We believe that it is time to move toward a more complete harmonization of risk assessment approaches for cancer and non-cancer effects. Although the draft EPA guidelines for cancer risk assessment (EPA 1994b) point in this direction, they do not, in our opinion, go far enough. As argued above, separate approaches for carcinogens and non-carcinogens are not justified by the scientific evidence. More importantly, this dichotomy between cancer and non-cancer risk policies likely produces severe imbalances in the cost of regulation and the level of protection afforded by various regulatory decisions. Moreover, the current risk assessment procedure for carcinogens blurs the distinction between what is known (observable dose region and qualitative mechanisms of action) and what is not known (the shape of the actual dose-response curve in the low-dose region), which makes the process subject to misunderstanding and misuse. With the non-cancer methodology, on the other hand, incomplete articulation of the interpretation of certain uncertainty factors makes it difficult to devise research strategies to improve the process or to utilize a broader data base in arriving at exposure standards. We advocate a common framework for setting acceptable human exposures as the default for both cancer and non-cancer that incorporates and adapts some features from each of the current dual approaches.
Because of the inherent uncertainties in estimates of low-dose human risks -- particularly when based upon animal data -- the recommended approach would not involve quantitative low-dose extrapolation of animal data. Instead, it would involve determining a risk-specific exposure in animals (i.e., NOAEL, ED10 or benchmark dose [Crump 1984, 1995; Allen et al. 1994]), use of pharmacokinetic principles and data to determine an equivalent human exposure (i.e., an exposure that would produce the same tissue concentration of the active toxin at the target site in humans) and application of an appropriate composite safety factor. The benchmark approach has several advantages over the NOAEL (Crump 1984, 1995) that we believe warrant its use whenever appropriate data are available. Multiple default procedures could be provided for conducting cross-species extrapolation in order to properly reflect the impact of route of administration and mechanism of toxicity on equivalent target tissue exposures in experimental animals and humans. In the optimal case, pharmacokinetic modeling would permit association of the observed effects with a measure of target tissue dose and extrapolation of tissue dose across species and among individuals in the human population. A single composite safety factor (or uncertainty factor) would then be applied, based on scientific judgement, that could reflect the severity of the endpoint, mechanistic information impacting the dose-response, human variability, and other uncertainties (e.g., inadequacy of database).
The recommended safety factor approach to risk assessment of both carcinogens and non-carcinogens is not based on a judgment that thresholds exist for most effects. Rather it is based on the conviction that low-dose risks remain largely unknown for both carcinogens and non-carcinogens, and that even the accuracy and/or relevance of the categorization as "carcinogen" or "non-carcinogen" is subject to doubt. Furthermore, we view the selection of the safety factor as involving, not a scientific judgment as to where a threshold lies, but a combination of scientific judgment about low-dose risks and, perhaps more importantly, policy considerations regarding how much safety is appropriate. Although our proposed method would not provide quantitative estimates of low-dose risks, its adoption as a default does not preclude the development of such estimates when warranted.
References
Allen B, Kimmel C, Kavlock R, Faustman E. 1994. Dose-response assessment for development toxicity. II. Comparison of generic benchmark dose estimates with NOAELs. Fund Appl Toxicol 23:487-495.
Andersen ME, Mills JJ, Jirtle RJ, and Greenlee WF. 1995. Negative selection in hepatic tumor promotion in relation to cancer risk assessment. Toxicology 102: 233-237.
Beck BD, Conolly RB, Dourson ML, Guth D, Hattis D, Kimmel C, and Lewis SC. 1993. Improvements in quantitative noncancer risk assessment. Fundam. Appl. Toxicol. 20:1-14.
Crump K. 1984. A new method for determining allowable daily intakes. Fund Appl Toxicol 4:854-871.
Crump K. 1995. Calculation of benchmark doses from continuous data. Risk Anal 15:79-89.
Crump K, Hoel D, Langley C and Peto R. 1976. Fundamental carcinogenic processes and their implications to low dose risk assessment. Cancer Res 36: 2973-2979.
Crump K, Haseman J, Krewski D and Wang Y. 1996. Estimates of the number of liver carcinogens in bioassays conducted by the National Toxicology Program. Abstract for the Annual Meeting of the Society of Toxicology, Anaheim, California, March 10-14, 1996.
Environmental Protection Agency (EPA). 1992. Request for comments on draft report of cross-species scaling factor for cancer risk assessment. Fed. Reg. 57 (1992) 24152.
Environmental Protection Agency (EPA). 1994a. Health assessment Document for 2,3,7,8 Tetrachlorodibenzo-p-Dioxin (TCDD) and Related Compounds. EPA/600/BP-92/001.
Environmental Protection Agency (EPA). 1994b. Draft revisions to the guidelines for cancer risk assessment. EPA/600/BP-92/003.
Larson JL, Wolf DC and Butterworth BE. 1994. Induced cytotoxicity and cell proliferation in the hepatocarcinogenicity of chloroform in female B6C3F1 mice: comparison of administration by gavage in corn oil vs. ad libitum in drinking water. Fundam Appl Toxicol 22:90-102.
National Research Council (NRC). 1977. Safe Drinking Water and Health. National Academy of Sciences, Washington, DC.
Reitz RH, Mendrala AL, Corley RA, Quast JF, Gargas ML, Andersen ME, Staats DA, and Conolly RB. 1990. Estimating the risk of liver cancer associated with human exposures to chloroform using physiologically based pharmacokinetic modeling. Toxicol. Appl. Pharmacol. 105:443-459. |
The Rodent High-Dose Cancer Test is Limited at Best: When Cell Division is Ignored, then Risk Assessment will be Flawed
Bruce N. Ames* and Lois Swirsky Gold*Ý
*Division of Biochemistry and Molecular Biology, University
of California, 401 Barker Hall, Berkeley, CA 94720-3202
Tel: (510) 642-5156; Fax: (510) 643-7935
ÝLife Sciences Division, Lawrence Berkeley Laboratory
Berkeley, CA 94720; http://potency.berkeley.edu/cpdb.html
As currently conducted, rodent bioassays provide inadequate data to estimate human risk at low dose. Accumulating evidence suggests that chronic cell division is a major reason why half the chemicals tested are carcinogenic. To the extent that such a high-dose effect contributes to positivity, humans are likely exposed to a huge background of rodent carcinogens and linear extrapolation to low dose is inappropriate.
The importance of cell division in mutagenesis and carcinogenesis.
Endogenous DNA damage from normal oxidation is enormous. Considerable evidence suggests that oxidative damage is a major factor, not only in aging, but in the degenerative diseases of aging such as cancer. The steady-state level of oxidative damage in DNA is over one million oxidative lesions per rat cell[1]. Thus, from first principles, the cell division rate must be a factor in converting lesions to mutations and thus cancer[2]. Raising the level of either DNA lesions or cell division will increase the probability of cancer. Just as DNA repair protects against lesions, p53 guards the cell cycle and protects against cell division if the lesion level gets too high; however neither defense is perfect[3]. Cell division is also a major factor in loss of heterozygosity through non-disjunction and other mechanisms[4, 5]. The critical factor is chronic cell division in stem cells, not in cells that are discarde
Half the chemicals tested in rodents are carcinogens.
Chronic cell division is plausible as the major reason that more than half the chemicals are classified as carcinogens when tested at the maximum tolerated dose (MTD) in standard rodent cancer bioassays[2, 5, 6]. Of the chemicals tested in both rats and mice, 60% are positive; even among the known non-mutagens, 49% are carcinogenic (among the mutagens, 78% are carcinogenic)[6]. The high positivity rate is consistent for synthetic chemicals, natural chemicals (99.9% of the chemicals humans are exposed to are natural) [6-8] natural pesticides, chemicals in roasted coffee, and the proportion positive has not changed through the years of testing[5, 9]. Half the drugs in the Physician's Desk Reference that report animal cancer test results are carcinogenic [10]. The Innes series of tests in l969 of 119 synthetic chemicals, mainly all of the commonly used pesticides of the time, is frequently cited as evidence that the proportion of carcinogens in the world of chemicals is low, as only 9% were judged positive. We have pointed out that these tests were quite deficient in power compared to modern tests [9, 11], and we have now reanalyzed Innes by asking whether any of the Innes-negative chemicals have been retested using current protocols. We found that 34 have been retested and 16 were carcinogenic, again about half [11].
Cell division and the high positivity rate in bioassays.
What is the explanation for the high positivity rate in high-dose animal cancer tests? We have rejected bias in picking more suspicious chemicals as the major explanation for the results for numerous reasons [9, 11]. One explanation for a high positivity rate that is supported by an ever increasing array of papers is that the MTD of a chemical can cause chronic cell killing and cell replacement in the target tissue, a risk factor for cancer that can be limited to the high dose. Thus it seems likely that a high proportion of the chemicals in the world may be "carcinogens" if run through the standard rodent bioassay at the MTD, but this will be primarily due to the effects of high doses for the non-mutagens, and a synergistic effect of cell division at high doses with DNA damage for the mutagens [2, 5, 12]. Ad libitum feeding in the standard bioassay can also contribute to the high positivity rate [13, 14], plausibly by increased cell division due to high caloric intake [2, 14].
Correlation between cell division and cancer.
Many studies on rodent carcinogenicity show a correlation between cell division at the MTD and cancer. Cunningham has analyzed 15 chemicals at the MTD, 8 mutagens and 7 non-mutagens, including several pairs of mutagenic isomers, one of which is a carcinogen and one of which is not [15-23]. He found a perfect correlation between cancer causation and cell division in the target tissue: the 9 chemicals causing cancer caused cell division in the target tissue and the 6 chemicals not causing cancer did not. A similar result has been found in the analyses of Mirsalis [24], e.g. both dimethyl nitrosamine (DMN) and methyl methane sulfonate (MMS) methylate liver DNA and cause unscheduled DNA synthesis, but DMN causes both cell division and liver tumors, while MMS does neither. A recent study on the mutagenic dose response of the carcinogen ethylnitrosourea concludes that cell division is a key factor in its mutagenesis and carcinogenesis [25]. Chloroform at high doses induces liver cancer by chronic cell division [26]. Extensive reviews on rodent studies [2, 5, 27-30] document that chronic cell division can induce cancer. There is also a large epidemiological literature reviewed by Preston-Martin, Henderson and colleagues [31, 32] showing that increased cell division by hormones and other agents can increase human cancer.
Risk Assessment.
To use bioassay results for risk assessment requires two extrapolations: a qualitative inter-species extrapolation from rats or mice to humans, and a quantitative extrapolation from high to low dose. Indirect evidence can be used to validate the qualitative species extrapolation: how concordant are carcinogenicity results in rats and mice, two closely related species tested under similar conditions? Seventy-five percent of test chemicals give the same positivity results, i.e. concordance, in rats and mice. Our recent analysis, however, indicates that an observed concordance of 75% can arise if the true concordance is anything between 20% and 100%, and in particular observed concordance can seriously overestimate true concordance [33]. This provides little confidence in inter-species extrapolation. Moreover, the good correlation observed in carcinogenic potency between rats and mice has been shown to be primarily an artifact due to the correlation of toxicity (MTD) in the two species and the fact that potency estimates are constrained to a narrow range about the MTD.
In regulatory policy, the "virtually safe dose" (VSD) is estimated from bioassay results using a linear model. To the extent that carcinogenicity in rodent bioassays is due to the effects of high doses for the non-mutagens, and a synergistic effect of cell division at high doses with DNA damage for the mutagens, then this model is inappropriate. Moreover, as currently calculated, VSD can be known without ever conducting a bioassay: for 96% of the NCI/NTP rodent carcinogens, the VSD is within a factor of 10 of the ratio MTD/740,000 [34]. This is about as precise as the estimate obtained from conducting near-replicate tests of the same chemical [34]. Conclusion.
Taking cell division into account will make priority setting in cancer prevention more effective [12]. For example, regulatory policy aimed at reducing tiny exposures to synthetic rodent carcinogens [8] has confused the public about what factors are important for preventing cancer [3], and has diverted resources from more important health risks [3, 11, 35].
References
| 1. | Ames, B.N., M.K. Shigenaga, and T.M. Hagen, Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA, 1993. 90: p. 7915-7922. |
| 2. | Ames, B.N., M.K. Shigenaga, and L.S. Gold, DNA lesions, inducible DNA repair, and cell division: Three key factors in mutagenesis and carcinogenesis. Environ. Health Perspect., 1993. 101(Suppl 5): p. 35-44. |
| 3. | Ames, B.N., L.S. Gold, and W.C. Willett, The causes and prevention of cancer. Proceedings of the National Academy of Science USA, 1995. 92: p. 5258-5265. |
| 4. | Vomiero-Highton, G. and J. Heddle, An assay for loss of heterozygosity in vivo at the Dlb-1 locus. Mutagenesis, 1995. 10(5): p. 381-384. |
| 5. | Ames, B.N. and L.S. Gold, Chemical carcinogenesis: Too many rodent carcinogens. Proc. Natl. Acad. Sci. USA, 1990. 87: p. 7772-7776. |
| 6. | Gold, L.S., et al., The fifth plot of the Carcinogenic Potency Database: Results of animal bioassays published in the general literature through 1988 and by the National Toxicology Program through 1989. Environ. Health Perspect., 1993. 100: p. 65-135. |
| 7. | Ames, B.N., M. Profet, and L.S. Gold, Dietary pesticides (99.99% all natural). Proc. Natl. Acad. Sci. USA, 1990. 87: p. 7777-7781. |
| 8. | Gold, L.S., et al., Rodent carcinogens: Setting priorities. Science, 1992. 258: p. 261-265. |
| 9. | Gold, L.S., et al., Interspecies extrapolation in carcinogenesis: Prediction between rats and mice. Environ. Health Perspect., 1989. 81: p. 211-219. |
| 10. | Davies, T.S. and A. Monro, Marketed Human Pharmaceuticals Reported to be Tumorigenic in Rodents. Journal of the American College of Toxicology, 1995. 14(2): p. 90-107. |
| 11. | Ames, B.N. and L.S. Gold, eds. The Causes and Prevention of Cancer: Gaining Perspectives on Management of Risk. Improving Risk Management: From Science to Policy, ed. R.W. Hahn. 1996, Oxford University Press, in press. |
| 12. | Butterworth, B., R. Conolly, and K. Morgan, A strategy for establishing mode of action of chemical carcinogens as a guide for approaches to risk assessment. Cancer Letters, 1995. 93: p. 129-146. |
| 13. | NTP (National Toxicology Program), Effect of dietary restriction on toxicology and carcinogenesis studies in F344/N rats and B6C3F1 mice. Technical Report Series No. 460: Research Triangle Park, N.C.: U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, 1995. |
| 14. | Hart, R., D. Neumann, and R. Robertson, eds. Dietary restriction: Implications for the design and interpretation of toxicity and carcinogenicity studies. . 1995, ILSI Press: Washington, D.C. |
| 15. | Cunningham, M.L., et al., Correlation of hepatocellular proliferation with hepatocarcinogenicity induced by the mutagenic noncarcinogen: Carcinogen pair--2,6-and 2,4-diaminotoluene. Toxicol. Appl. Pharmacol., 1991. 107: p. 562-567. |
| 16. | Cunningham, M.L. and H.B. Matthews, Relationship of hepatocarcinogenicity and hepatocellular proliferation induced by mutagenic noncarcinogens vs. carcinogens. II. 1- vs. 2-nitropropane. Toxicol. Appl. Pharmacol., 1991. 110: p. 505-513. |
| 17. | Cunningham, M.L., M.R. Elwell, and H.B. Matthews, Site-specific Cell Proliferation in Renal Tubular Cells by the Renal Tubular Carcinogen tris (2,3-Dibromopropyl)phosphate. Environ. Health Perspect., 1993. 101(Suppl 5): p. 253-258. |
| 18. | Cunningham, M.L., M.R. Elwell, and H.B. Matthews, Relationship of carcinogenicity and cellular proliferation induced by mutagenic noncarcinogens vs carcinogens. Fundam. Appl. Toxicol., 1994. 23: p. 363-369. |
| 19. | Cunningham, M.L., et al., Early responses of the liver of B6C3F1 mice to the hepatocarcinogen oxazepam. Toxicol. Appl. Pharmacol., 1994. 124: p. 31-38. |
| 20. | Yarbrough, J., et al., Carbohydrate and oxygen metabolism during hepatocellular proliferation: A study in perfused livers from mirex-treated rats. Hepatology, 1991. 13: p. 1229-1234. |
| 21. | Cunningham, M.L., et al., The hepatocarcinogen methapyrilene but not the analog pyrilamine induces sustained hepatocellular replication and protein alterations in F344 rats in a 13-week feed study. Toxic. and Appl. Pharmacol., 1995. 131: p. 216-223. |
| 22. | Thottassery, J., et al., Regulation of perfluorooctanoic acid -- induced peroxisomal enzyme activities and hepatocellular growth by adrenal hormones. Hepatology, 1992. 15(2): p. 316-22. |
| 23. | Hayward, J., et al., Differential in vivo mutagenicity of the carcinogen/non-carcinogen pair 2,4- and 2,6 diaminotoluene. Carcinogenesis, 1995. 16(10): p. 2429-2433. |
| 24. | Mirsalis, J.C., Provost, G.S., Matthews, C.D., Hamner, R.T., Schindler, J.E., O'Loughlin, K.G., MacGregor, J.T. and Short, J.M., Induction of hepatic mutations in lacI transgenic mice. Mutagenesis, 1993. 8(3): p. 265-271. |
| 25. | Shaver-Walker, P., et al., Enhanced somatic mutation rates induced in stem cells of mice by low chronic exposures. Proc. Natl. Acad. Sci. USA, 1995. 92(25): p. 11470-11474. |
| 26. | Larson, J., D. Wolf, and B. Butterworth, Induced cytotoxicity and cell proliferation in the hepatocarcinogenecity of chloroform in female B6C3F1 mice: comparison of administration by gavage in corn oil vs ad libitum in drinking water. Fundam. Appl. Toxicol., 1994. 22(1): p. 90-102. |
| 27. | Cohen, S. and T. Lawson, Rodent bladder tumors do not always predict for humans. Cancer Letters, 1995. 93: p. 9-16. |
| 28. | Cohen, S.M. and L.B. Ellwein, Genetic errors, cell proliferation, and carcinogenesis. Cancer Res., 1991. 51: p. 6493-6505. |
| 29. | Cohen, S., Human Relevance of Animal Carcinogenicity Studies. Regul. Toxicol. Pharmacol., 1995. 21: p. 75-80. |
| 30. | Counts, J. and J. Goodman, Principles underlying dose selection for, and extrapolation from, the carcinogen bioassay: dose influence mechanism. Reg. Toxic. Pharm., 1995. 21: p. 418-421. |
| 31. | Preston-Martin, S., et al., Increased cell division as a cause of human cancer. Cancer Res., 1990. 50: p. 7415-7421. |
| 32. | Preston-Martin, S., et al., Spinal meningiomas in women in Los Angeles County: Investigation of an etiological hypothesis. Cancer Epidemiol. Biomarkers, 1995. 4: p. 333-339. |
| 33. | Lin, T., L.S. Gold, and D. Freedman, Bias in qualitative measures of concordance for rodent carcinogenicity test. Technical Report No. 439: Berkeley, C.A.: Department of Statistics, University of California, 1996. |
| 34. | Gaylor, D.W. and L.S. Gold, Quick Estimate of the Regulatory Virtually Safe Dose Based on the Maximum Tolerated Dose for Rodent Bioassays. Regul. Toxicol. Pharmacol., 1995. 22: p. 57-63. |
| 35. | Tengs, T.O., et al., Five-hundred life-saving interventions and their cost-effectiveness. Risk Anal., 1995. 15(3): p. 369-390. |
The Cancer Risk Assessment Paradigm An Experimentalist's Viewpoint Lucy M. Anderson, Ph.D.
Chief, Perinatal Carcinogenesis Section, Laboratory of
Comparative Carcinogenesis, Division of Basic Sciences,
National Cancer Institute, FCRDC, Frederick, MD
Tel: (301) 846-5600; Fax: (301) 846-5946
Is the simple, linear-extrapolation, no dose-no cancer approach to cancer risk assessment still the best strategy? If not, then what?
This approach has appeal in its simplicity and conservatism. In theory, it ensures protection of the populace against any identified hazard. Its weaknesses would seem to be at least two. It will often be inaccurately overconservative, resulting in unnecessary cost of containment, and inappropriate curtailment of freedom of production and use of beneficial substances. Worse, it will often if not usually be unenforceable, in a society which emphasizes freedom and has shrinking resources for policing of exposures. In attempting to do the impossible extreme, the strategy may result in less protection than would rigorous control of a more limited number of substances certain to be dangerous even at low doses.
The obvious alternative is to base risk assessment on a thorough understanding of the mechanisms underlying risk. Given adequate information, we could accurately and in detail estimate the cancer risks, for types of cancer, of a subgroup of individuals, and then carry out rational risk/benefit analysis to choose candidates for serious and effective enforcement. Is this a realistic prospect?
Human cancer risk is the net outcome of a time continuum of situations, starting even before conception, each comprised of a complexity of factors: toxic chemicals/radiation; chemicals interacting positively or negatively with the toxicants or the biological substrate; viruses and bacteria; host features such as genetics, hormones, and immune status; tissue specific phenomena; diet; lifestyle aspects; and so forth. Genotoxic events which contribute to both the initiation and progression of neoplasms, and nongenotoxic or epigenetic factors that encourage or suppress tumor development, are all undoubtedly important. Although considerable basic and descriptive information has accumulated in recent years on certain sectors of this tapestry, such as carcinogen activation and DNA damage, mutation, and altered oncogenes and tumor suppressor genes in human cancers, we are still far away from a sufficiently comprehensive understanding of process, to permit a detailed assessment of cancer risk based on specifics. Olden and Klein1 presented last year a particularly excellent overview of some of the information still needed at the molecular, cellular, and organismal level in order "to design effective prevention and intervention strategies".
The complexity in risk, both linear with regard to timeframe and cross-sectional with regard to exposure components, is daunting. It will eventually be mastered only by integration of molecular and cellular understanding with careful, thorough modeling in whole-animal, whole-life contexts. In direct contrast to this need, fashion in cancer research, enforced by funding decisions, has become ever more reductive and molecular. Senior leaders can be heard suggesting that animal research should be abandonned altogether, to concentrate on molecular analysis of human material. This is nonsense. Even full understanding of the structure of the human genome, the intricacies of the cell cycle, and the mysterious dynamics of intracellular signaling, and discovery of all possible human oncogenes and tumor suppressor genes, exciting as these accomplishments will be, will not together provide us with a detailed description of human disease risk. This basic molecular and cellular information will need to be put together with all of the other variables, in the biological context of the whole animal and the whole lifetime, before useful answers will be forthcoming.
Obviously, to meet these needs, animal tumor endpoint experimentation needs to go far beyond the treat-with-a-high-dose-and-count-the-tumors bioassay approach. Work is needed that addresses risk factors and mechanisms in a specific and up-to-date way. Along these lines, studies that are currently popular include analysis of carcinogen-induced tumors for mutated oncogenes and tumor suppressor genes, use of transgenic and gene knock-out mice, and modulation of outcome by diet and putative tumor preventative substances. Even these projects represent a small minority of published cancer research effort. In 1995, of approximately 950 papers published in 24 issues of Cancer Research, 29 (3%) could be described as modeling human cancer risk in animals with reference to a neoplastic or preneoplastic endpoint. The more specialized Carcinogenesis had a better average, with 22/90 (24%) of papers in the first three issues. Five 1995 issues taken at random of the prestigious Journal of the National Cancer Institute had 0/34 articles of this sort.
The following are some suggestions for administrative and scientific tactics, related to whole-animal experimentation, that might bring us more rapidly to the point where we can perform detailed and accurate, knowledge-based risk assessment and management.
| (1) | Attract more excellent scientists at all levels to participate in this sort of endeavor, with the enticement of funding. This is particularly important for outstanding younger scientists, almost all of whom, these days, want to learn molecular biology. This could be done with RFAs that call for expertise in both tumor endpoint work and some important risk-related molecular parameter. Funding should be sufficient in amount and duration to permit tumor outcome analysis. A special Study Section might be needed to review these applications, in view of the uncommonness of this type of research at present. |
| (2) | Encourage more active dialogue between experimentalists and epidemiologists. Modeling of human cancer risk in animals requires detailed understanding of the current consensus on the status and meaning of epidemiological findings. Similarly, epidemiologist should be testing current risk concepts that are based on a thorough understanding of carcinogenesis. An idea for fostering this interaction might be a series of Hypothesis papers coauthored by an experimentalist and an epidemiologist. Journal editors might issue invitations to specific pairs of individuals for such efforts, with good effect. Another idea is to supply study sections that evaluate epidemiology with experimentalists, and those that deal with carcinogenesis-related experimental proposals with epidemiologists. One or two senior people of broad experience as permanent members, plus a Reviewers Reserve for specific ad hoc duties, would suffice. |
| (3) | Experimentally, attend to some facets of risk that are almost certainly important, but currently neglected, largely or entirely. |
| (a) | Stage-of-life susceptibilities. Individuals are likely to be more sensitive to carcinogens, and to tumorigenesis-modulating effects, early in life, during the perinatal period, and late in life, during the beginning of the senescence process. Epidemiology reveals, over and over again, that early-life situations have a large influence on adult cancer risk, e.g., breast cancer2. Some attention is given to control of and evaluation of the effects of transplacental exposure, but exposure of infants and children, who are likely to be more susceptible than fetuses, is only recently being given special focus, with some alarming results. A new study of inner city children revealed strikingly high exposure to environmental tobacco smoke3 and benzene4. Evidence is mounting that exposures of fathers may have significant and varied impact on risk of cancers in their children.5 Yet laboratory experimentation being reported on transplacental and preconception carcinogenesis is minimal, and virtually absent for the immature young. At the other end of life, events occurring in humans in their 50's, early in the aging process, may modulate tumor outcome in their 60's and 70's, but animal modeling of such phenomena is rare to nonexistent. |
| (b) | Whole-animal phenomena. The roles at the organismal level of the immune and neuroendocrine systems, and cytokines and growth factors are correctly emphasized by Olden and Klein1. These are high-fashion areas being vigorously pursued at the cellular level. However, good studies on these topics, at the organismal level and relating specifically to a tumor endpoint, are scarce indeed. Another example may be taken from our work. Alcohol, a strong human cancer risk factor but not a carcinogen in animals, potentiates tumorigenesis in rodents due to nitrosamines. Research on cellular level phenomena has revealed 2- to 4-fold effects of ethanol on possibly relevant parameters such as carcinogen activation, repair of DNA damage, and cell proliferation.6 By contrast, ethanol in vivo causes much larger increases, 10- to 20-fold, in nitrosamine tissue exposure, DNA adducts, and tumors in monkeys and mice, simply by inhibiting hepatic clearance of the carcinogen. This humble whole-animal toxicokinetic effect is sufficient to account for observed increases in human risk, whereas the more glamorous cell-based changes are not. |
| (c) | Dose effects. Humans are in general exposed to quite low doses of chemicals and radiation, usually chronically or intermittently. There is evidence that critical events, such as oncogene activation, are altered qualitatively as dose to animals is lowered.7,8 This is obviously a topic that demands systematic study. The time for mega-mouse exposure studies is past, but new, highly sensitive techniques for detecting very low levels of critical alterations such as DNA adducts and mutations can now be brought to bear. |
| (4) | Place more emphasis on diet-related experimentation. Epidemiological studies show major effects of diet, positive and negative, on human cancer risk, and chemoprevention trials have been undertaken based on these findings, with mixed results. Yet relatively few laboratories are carrying out related experimentation with animals. Such work is long, laborious, and expensive. It will need specific encouragement. |
| (5) | Beware of the strait-jacketing effects of fashion. Nitrosamine and polycyclic aromatic hydrocarbon carcinogens, high-profile topics in a previous era, are now studied experimentally by only the dogged and the stubborn, and then often as gene-damaging tools rather than risk agents. Yet evidence implicating both of these chemical classes in human cancer etiology has continued to mount. Experimentation with transgenic and gene knock-out mice has become popular, and has exciting potential, but it must be remembered that these are not normal organisms. Conclusions from such animals cannot be assumed to be generally valid, until tested in other contexts. |
| (6) | Utilize and develop more human-relevant animal models. Expression of human genes as transgenes in rodents is one approach to this, but has the limitation noted above. Systematic screening could be undertaken of mouse strains for polymorphisms thought to be linked to human cancer risk, for example, those in certain cytochrome P450 and glutathione S transferase genes. Of course, rodents will remain rodents, regardless of their specific engineering, greatly separated from the human by size and phylogenetic development, so nonhuman primate models could be utilized in a selective way for specific hypothesis testing. |
References
| (1) | K. Olden and J.-L. Klein. Environmental health science research and human risk assessment. Mol. Carcinogenesis 14: 2-9, 1995. |
| (2) | G.A. Colditz and A.L. Frazier. Models of breast cancer show that risk is set by events of early life: prevention efforts must shift focus. Cancer Epidemiol. Biomark. Prevent. 4: 567-571, 1995. |
| (3) | V. Weaver, C. Davoll, S.E. Murphy, J. Sunyer, P.J. Heller, A. Fitzwilliam, S. Colosimo and J.D. Groopman. Environmental tobacco smoke exposure in inner-city children. Cancer Epidemiol. Biomark. Prevent., in press, 1996. |
| (4) | V. Weaver, C. Davoll, P.J. Heller, A. Fitzwilliam, H. Peters, J. Sunyer, S. Murphy, S.S. Hecht, G. Goldstein and J.D. Groopman. Benzene exposure, assessed by urinary trans, trans-muconic acid, in urban children with elevated blood lead levels. Environ. Health Perspect., in press, 1996. |
| (5) | G.R. Bunin, K. Noller, P. Rose and E. Smith. Carcinogenesis. In Occupational and Environmental Reproductive Hazards: A Guide for Clinicians. M. Paul, Ed. Williams and Wilkins, Baltimore, 1992. |
| (6) | S.K. Chhabra, V.L. Souliotis, S.A. Kyrtopoulos, and L.M. Anderson. Nitrosamines, alcohol, and gastrointestinal tract cancer: recent epidemiology and experimentation. In Vivo, in press, 1996. |
| (7) | B. Chen, L. Liu, A. Castonguay, R.R. Maronpot, M.W. Anderson and M. You. Dose-dependent ras mutation spectra in N-nitrosodiethylamine induced mouse liver tumors and 4 (methylnitrosamino)-1-(3-pyridyl)-1-butanone induced mouse lung tumors. Carcinogenesis 14: 1603-1608, 1993. |
| (8) | S. Manam, G.A. Shinder, D.J. Joslyn, A.R. Kraynak, C.L. Hammermeister, K.R. Leander, D.J. Ledwith, S. Prahalada, M.J. van Zwieten and W.W. Nichols. Dose-related changes in the profile of ras mutations in chemically induced CD-1 mouse liver tumors. Carcinogenesis 16: 1113-1119, 1995. |
Is There a Need for a New CancerRisk Assessment Paradigm?
Robert T. Drew
American Petroleum Institute, HESD, 1220 L Street, Northwest
Washington, DC 20005-4070; Tel: (202) 682-8308
Do we need a new paradigm? Probably not. What we do need is a more judicious application of the current paradigm. I am concerned that if we stop using the bioassay there is no guarantee that the replacement paradigm will perform any better. In this paper I want to address four issues: the original concept of the Maximum Tolerated Dose (MTD) and how it has changed; our failure to apply new scientific developments to that concept; the misuse of data derived from studies where the MTD was clearly exceeded; and finally, the concept of regulating for public health vs. individual risk. The cancer bioassay was developed in order to determine if chronic exposure to a particular chemical could cause cancer in the absence of other toxic effects. Exposures were carried out at the MTD and some fraction (usually half) of the MTD. The MTD was defined as a dose which would produce no overt toxicity to any organ other than a slight (10%) weight loss (actually a decrease in weight gain) of the animals under test. Originally the weight loss "could" be observed. Gradually "could" turned into "should", and the observation of weight loss was soon considered necessary to document that an MTD was being used. The rationale for not causing overt toxicity was based on the notion that, if a chemical caused overt toxicity, it would be noticed and exposures would be stopped because of the observed effect. The question was whether chronic exposure to a chemical that otherwise was not toxic could cause cancer.
In the 30 or so years we have been performing bioassays, toxicology has advanced markedly and we have much better tools to define toxic effects. In many cases slight alteration of normal homeostasis can lead to toxicity. For example, several chemicals increase the occurrence of thyroid tumors secondary to microsomal enzyme induction which increases clearance of thyroid hormone. Pharmacokinetics studies allow us to measure a variety of indices which, if markedly changed, should be considered a toxic effect. The best example of this might be pulmonary clearance of particles, e.g. titanium dioxide. It is now recognized that the lung has a finite ability to handle particles and at some point, particle clearance will be decreased. In this case, the modification of concentration-time relationships can drastically alter experimental results, e.g. two to three fold increases in exposure concentration could result in dose-to-target-tissue increases of ten to twenty fold. Another example is the induction of a secondary metabolic pathway which does not become active until the primary pathway is saturated. If we know homeostasis has been altered, or pulmonary clearance has been impaired, or secondary mechanisms are being induced, these effects should be considered toxic effects and the MTD adjusted downward accordingly. The definition of "overtly toxic" has failed to keep pace with the developing science of toxicology.
The cancer bioassay was developed before risk assessment paradigms became popular. The bioassay was originally a safety evaluation paradigm with only two doses being tested. As we moved into risk assessment the bioassay added a third dose in order to better characterize dose response. If the data support the idea that the highest dose selected met the criteria for MTD, such risk assessments are probably appropriate. The problem occurs when the MTD is exceeded. When overt toxicity is noticed during a bioassay the study is usually completed as designed. The resultant cancer incidence data are used for risk assessment even when acute organ toxicity was observed, and mortality incidences required early termination of the study. Performing such calculations when the MTD was clearly exceeded is not appropriate. A current example of this is the use of data on the carcinogenicity of methyl tertiary butyl ether (MTBE) for risk assessment. In this case the lowest dose 400 ppm was a "no observed adverse effect level. The intermediate dose (3000 ppm) and the high dose (8000 ppm) both produced clear toxicity with both groups of male rats being sacrificed prior to the planned two year duration of the study. Thus, MTD was clearly exceeded yet some groups are using this data to calculate cancer risks associated with exposure to MTBE.
Finally, we are so enamored of the risk assessment models that we are becoming victims of numbers. The use of so-called risk "brightlines" (e.g. 10-6, 10-5 levels of risk) to establish compliance tests, or level of cleanup, was perhaps a consequence of our almost mystical faith in numbers, especially since the process producing them appears so sophisticated. Unfortunately, quantitative risk assessments have been used principally to estimate individual rather than population risks. Population risk is the domain of public health, while individual risk may have been derived from the tort process. In many cases, individual risk, not population risk, is the engine driving the environmental regulatory requirements. Thus expensive fixes are required in instances where fenceline concentrations of a pollutant may barely exceed a hypothetical individual risk of one-in-a-million while the population risks are indeed trivial (e.g. one calculated excess cancer death in the impacted population over several hundred years). For example, using EPA s potency factor for benzene and a population risk level of 10-6 leads to an environmental standard of 37 parts per trillion for benzene. Indeed, several states have adopted such a standard in spite of the fact that ambient levels of benzene usually range between 1 and 5 parts per billion. Risk managers must learn to ask the risk assessors about the estimated public health impact that an industry or a pollutant poses, rather than focusing exclusively on individual risk levels. Risk assessors must learn to provide these estimates of population risks whether or not they are requested.
We have to abandon this idea of protecting the population to a lifetime risk of one-in-a-million (10-6). Risks in the order of 10-6 include being struck by lightening (3.5x10-5) or being hit by falling airplanes (4x10-6). These are involuntary risks which the public clearly accepts. Voluntary risks that the public readily accepts include cigarette smoking and riding in automobiles. Approximately fifty thousand people die each year in automobile accidents. Thus the annual risk for the United States population of about 260 million is about one in 5200. The voluntary lifetime risk for a median lifetime of 72 years from automobiles is about one-in-75. Thus the United States public readily accepts voluntary lifetime risks of less than one-in-a-hundred. Perhaps we should consider a factor of 10 or even one-hundred between voluntary and involuntary risks. This would suggest 10-3 or 10-4 are acceptable levels of involuntary risk.
This country cannot afford to protect individuals to a lifetime risk of 10-6. A lifetime risk of 10-6 in the United States population of 260 million people equals approximately four deaths per year in a population experiencing about four million deaths/year of which about 25% or 1.0 million deaths per year are attributable to cancer. The majority of these cancers are hereditary or are related to lifestyle or dietary factors. These latter causes of cancer clearly fall into the category of voluntary risks. Clearly if we are concerned about cancer, we should put our money into prevention and cure, not into costly and ineffective pollution control devices. If the goal of environmental regulation is to reduce the overall death rate or improve the health of the population, regulating chemical risks to 10-6 will do very little toward achieving that goal.
In summary,
| € | We need better definition of the MTD using more sensitive indices of toxicity to define concentrations for bioassays; |
| € | Cancer risk assessments using data from studies where organ toxicity was observed need to be viewed with some skepticism; |
| € | The regulatory community needs to recognize that protecting the public to involuntary risks of 10-6 doesn t make sense when the public routinely accepts voluntary risks of less than 10-2. |
The Rodent Bioassay is Being Wrongly Used To Identify "Carcinogens".
A.M. Monro
Pfizer, Inc., Central Research Division, Eastern Point Road,
Groton, CT 06340; Tel: (203) 441-4853; Fax: (203) 441-4111
"Everything is a carcinogen?"
"Too many carcinogens." "Two million rodent carcinogens?"
These three pithy, provocative titles (1,2,3) highlight the morass into which the current paradigm for cancer risk assessment has led us. I shall address two themes that emerge from the titles: the word "carcinogen" and the ubiquity of these "carcinogens". Those who have to interpret the results of the currently mandated methods for the assessment of the carcinogenic activity of a chemical face a serious dilemma. Rodent carcinogenicity bioassays, normally carried out by exposing two species of rodents to the Maximum Tolerated Dose (MTD) of the chemical on a repeated daily basis for most of their life time (about 2 years) often result in a chemical being characterized as "a carcinogen". Why is this a problem?
Every text on the histopathological definition of neoplasia has biologically-meaningful criteria that allow a pathologist to diagnose and describe a tumor. While these criteria may still be evolving in some cases, they are essentially understood and accepted by pathologists. By contrast, there does not seem to be a meaningful definition of a "carcinogen". And the consequences of this are far reaching. A recent general text on toxicology describes a carcinogen as "an agent whose administration to previously untreated animals leads to a statistically significant increased incidence of neoplasms of one or more histogenic types as compared with the incidence in appropriate untreated animals" (4). The United States Biennual Report on Carcinogens (BRC) is required by Congress to compile a list of "substances which either are known to be carcinogens or which may reasonably anticipated to be carcinogensŠ" (5). The report explains that "carcinogens" are classified from data in humans, while those substances "anticipated" to be carcinogens are classified on the basis of either less convincing data from humans or from data from animals. Other schemes have been proposed to define a "carcinogen" (6). The International Agency for Research on Cancer (IARC) and the U.S. National Toxicology Program have largely avoided this semantic trap by not labeling agents as "carcinogens". Instead, the classification categories are based on the strength of the evidence for "carcinogenicity" of the agent in humans, animals, or laboratory test systems.
What is a "carcinogen"?
If one can define a "carcinogen", one might conclude that one can also define a "non-carcinogen", even if only by exclusion. As will be argued below this is equally meaningless. To label a chemical as a "carcinogen" implies that it possesses an inherent property of "carcinogenicity" that can be expressed in any species. [As an aside, I would point out that if this were so, a positive result for carcinogenicity in a rodent bioassay in any one species would identify a "carcinogen"]. The analysis of vast data bases derived from hundreds of chemicals indicate that the concordance between bioassay results from rats and mice is only about 70%. Many chemicals appear to exhibit carcinogenic activity only in one species or in one strain or in one sex and, after diligent investigation, we often elucidate plausible secondary mechanisms that can explain such specificity. This leads to a spectrum of "carcinogens" from "single site/sex/species carcinogens" through to "trans-species common-site carcinogens" (7). So where does that leave phenobarbitol and DDT, two "trans-(rodent) species carcinogens" that appear not to be carcinogenic to humans? In addition, the carcinogenic activity of a chemical may also depend on the route of administration e.g. parental vs. oral, or the rate of administration e.g. via oral gavage or via the drinking water. Finally, can one attach the label "carcinogen" to a chemical that increases the incidence of one type of tumor but decreases that of another, or that increases tumor incidence in one species but decreases it in another, or that increases tumor incidence in one sex but decreases it in the other? (8).
Repercussions of the label "carcinogen"
The word "carcinogen" has an emotive impact on all those outside the circle of carcinogenicity testing, and is particularly newsworthy in the hands of the media. A useful chemical stigmatized as a "carcinogen" may be banned or limited in use. The idea of a "carcinogen" being added to our food supply is abhorrent, which is ironic in light of the hundreds of naturally-recurring (rodent) "carcinogens" that we consume in our food daily (9,10). If we insist on identifying such chemicals as "carcinogens", then we should equally acknowledge the presence in food of hundreds of "anti-carcinogens". Several cumulative analyses indicate that rodent bioassays as carried out hitherto have produced a positive result for about half the chemicals tested (11,12,13). It has been suggested that in the current paradigm almost any chemical, if tested at sufficiently high a dose in a sufficiently sensitive strain of rodent for sufficiently long, may give a positive carcinogenic response and be labeled as a "carcinogen" with all the frightening connotations for human safety of such a label (14, 15). This is absurd.
What can and should be done?
Two possibilities present themselves. One can consider whether the bioassay is so defective and confusing that it should be abandoned in its present form or one can try to modify the bioassay to improve its relevance. I consider that the standard strains of rodents are so deeply flawed as surrogates for humans that their value is extremely limited for cancer risk assessment in humans. In addition to the inconsistency of response cited earlier (species-species, sex-sex, tumor increases/decreases), most of the strains used hitherto rarely get tumors, either spontaneous or chemically-induced, in some of the commonest sites of human cancer (colorectal, prostate, stomach) and, conversely, are highly susceptible to tumorigenicity in sites in which cancer is relatively rare in humans (liver, adrenal, testis and, tobacco apart, lung). Even if one could devise a relevant genetically-homogenous strain of rodent, it seems that phenotypical instability derived from variability in gene expression is always going to confound interpretation of the outcome (16). In summary, the outlook for even a patched-up bioassay is bleak.
Whither the rodent bioassay?
Now let me try to be positive yet, at the same time, pragmatic. Is it too naive to suggest that a strategy of detecting carcinogenic hazards of chemicals to humans should be based on the knowledge available on the few agents considered by IARC to be carcinogenic to humans? These agents all possess one or more of four properties: genotoxicity (in vivo), immune suppression, hormonal activity or chronic irritation/inflammation. These properties can be largely identified and characterized in the laboratory and by appropriate three month toxicology studies in animals. With human pharmaceuticals, this information can be complemented by studies in humans that seek, under relevant conditions of exposure, the presence of any risk factors identified in animals. This strategy, when taken together with the extent to which the outcome of rodent bioassays is now predictable, has led me to argue that the continued conduct of such studies is difficult to defend for human pharmaceuticals (17). For other chemicals, while the obtention of data from humans is often less feasible, it is nevertheless helpful to keep these risk factors prominently in mind when devising strategies to investigate, for example, identifiable groups of occupationally-exposed persons.
Is there a role for a patched up bioassay? A pragmatic improvement would be to have a much lower incidence of positive results provided, of course, that these were to include all those chemicals definitely relevant to human risk assessment. Davies and I have proposed that the use of an upper dose limit of 1000 mg/kg body weight (or dietary equivalent) and of shorter duration studies (15 - 18 months) would reduce in a useful manner the number of "rodent carcinogens" and, at the same time, still allow the detection of chemicals known to be carcinogenic to humans (18,19). Others consider that the major defect in the bioassay is that laboratory rodents overeat and are overweight, and that restriction of dietary intake leads to a greatly improved model (20). In the pharmaceutical field, the International Conference on Harmonisation has taken a step in the right direction by proclaiming in the Introduction to the Guideline on Dose Selection in Carcinogenicity Studies that the top dose should provide data that are "interpretable in the context of clinical use" (21). However, I consider that all such approaches are merely tinkering with the problem, given the intrinsic deficiencies of the standard strains of rodent.
A perspective on a future role for the bioassay
No one wishes to allow humans to be exposed to a chemical that is truly a carcinogenic hazard. But against the overall picture of the preventable causes of human cancer, is the rodent bioassay cost effective in identifying such hazards? The main preventable causes of human cancer are tobacco (30 - 40%), diet (20 - 40%), hormones, infections, diagnostic x-rays and life-style factors such as obesity, alcohol-containing beverages, and exposure to sunlight (9). The contribution from occupation exposure to chemicals has been estimated at no more than 4% and to pharmaceuticals at no more than 1%. Thus, the massive resources currently consumed by rodent bioassays are addressing only a minuscule fraction of the total human cancer load. The concept of developing rodent models genetically-modified to better replicate the susceptibility of humans to chemical carcinogenesis is a worthy objective, and it is being pursued vigorously (22,23). However, judgment on the utility of these transgenic models must be withheld, probably for several years. Perhaps their value will lie in obtaining a better assessment of whether chemicals that have evidence of genotoxicity that is either equivocal or obtained under unrealistically high exposure also exhibit carcinogenic activity under relevant conditions in vivo? I have an impression that genetic toxicologists still possess the MTD "mind-set" so prevalent in carcinogenicity testing until recently, and and are always seeking to maximize sensitivity. A considerable proportion of chemicals stigmatized as genotoxic probably do not portend a carcinogenic hazard, either to rodents or to humans. After all, about one third of the Salmonella-positive chemicals in the survey of 301 NTP chemicals were found not to be carcinogenic to rodents (24).
Conclusion
In the short term, it is probably a forlorn hope and quite unrealistic to appeal for the abandonment of the word "carcinogen" in its naked form. More useful would be to report evidence of carcinogenic activity, always coupled with three descriptors - species/strain, tumor type (location, malignancy, etc.) and exposure (intensity and duration). No longer would we have to agonize over whether a chemical "is" or "is not" (black and white categories) a carcinogen; instead we would have a scale of gray, ranging from no evidence of carcinogenicity under defined conditions, through to carcinogenicity observed in multiple organs at low dose levels with a rapid onset time. In the longer term, we should face the fact that the behemoth of the rodent bioassay is identifying hundreds of chemicals as "carcinogens" that do not contribute at all to human cancer. Its value is dwarfed into near insignificance when compared with the potential to reduce human cancer by public health policies aimed at changing the public perception of risk that emanates from the life style factors listed above.
References
| 1. | Higginson, J. (1987). Everything is a carcinogen. Regul. Toxicol. Pharmacol. 7:89-95. |
| 2. | Ames, B.N., Gold, L.W. (1990). Too many rodent carcinogens: mitogenesis increases carcinogenesis. Science. 249:970-971. |
| 3. | Ashby, J. (1994). Two million rodent carcinogens? The role of SAR and QSAR in their detection. Mut. Res. 305:3-12. |
| 4. | Pitot, H.C. and Dragan, Y.P. (1995). Chemical carcinogenesis. In: Casarett and Doull's Toxicology, the Basic Science of Poisons, Klaassen C.D. (ed.) Macmillan, NY. pp. 201-267. |
| 5. | Seventh Annual Report on Carcinogens (1994). U.S. Dept. HHS., National Toxicology Program, Research Triangle Park, NC 27709. |
| 6. | Ashby, J., Doerrer, N.G., Flamm, F.G., Harris, J.E., Hughes, D.H., Johannsen, F.R., Lewis, S.C., Krivanek, N.D., McCarthy, J.F., Mollenaar, R.J., Raabe, G.K., Reynnolds, R.C., Smith, J.M., Stevens, J.T., Teta, M.J., and Wilson, J.D. (1990). A scheme for classifying carcinogens. Regul. Toxicol. Pharmacol. 12:270-295. |
| 7. | Tennant, R.W. (1993). Stratification of rodent carcinogenicity bioassay results to reflect relative human hazard. Mutat. Res. 286:111-118. |
| 8. | Davies, T.S., Monro, A. (1994). The rodent carcinogenicity bioassay produces a similar frequency of tumor increases and decreases: Implications for risk assessment. Regul. Toxicol. Pharmacol. 20:281-301. |
| 9. | Ames, B.N., Gold, L.S., Willett, W.C. (1995). The causes and prevention of cancer. Proc. Natl. Acad. Sci. 92:5258-5265. |
| 10. | National Research Council (1996). Carcinogens and anticarcinogens in the human diet. Committee on Comparative Toxicity of Naturally Occurring and Synthetic Substances. National Academy Press, Washington, D.C. |
| 11. | Gold, L.S., Bernstein, L., Magaw, R. et al. (1989). Interspecies extrapolation in carcinogenesis: prediction between rats and mice. Environ. Health Perspect. 81:211-219. |
| 12. | Huff, J., Haseman, J., Rall, D. (1991). Scientific concepts, value, and significance of chemical carcinogenesis studies. Annu. Rev. Pharmacol. Toxicol. 31:621-52. |
| 13. | Davies, T.S., Monro, A. (1995). Marketed human pharmaceuticals reported to be tumorigenic in rodents. J. Amer. College Toxicol. 14:90-107. |
| 14. | Ashby, J., Purchase IFH (1993). Will all chemicals be carcinogenic to rodents when adequately evaluated? Carcinogenesis. 8:489-495. |
| 15. | Salsburg (1989). Does everything "cause" cancer: an alternative explanation of the "carcinogenesis" bioassay. Fundam. Appl. Toxicol. 13:351-358. |
| 16. | Wolff, G.L. (1996). Variability in gene expression and tumor formation within genetically homogenous animal populations in bioassays. Fundam. Appl. Toxicol. 29:176-184. |
| 17. | Monro, A. (1996). Are lifespan rodent carcinogenicity studies defensible for pharmaceutical agents? Exp. Toxicol. Pathol. 48:63-74. |
| 18. | Davies, T.S. and Monro, A.M. (1995). The case for an upper limit of 1000mg/kg in rodent carcinogenicity tests. Cancer Lett. 95:69-77. |
| 19. | Davies, T.S. and Monro, A.M. (1996). The duration of the rodent carcinogenicity bioassay necessary to detect agents carcinogenic to humans. Fundam. Appl. Toxicol. 30:Suppl., 201. |
| 20. | ILSI (International Life Sciences Institute). (1995). Dietary Restriction: Implications for the design and interpretation of toxicity and carcinogenicity studies (Hart, R., Neumann, D. and Robertson, R., eds). ILSI Press, Washington DC. Several papers. |
| 21. | International Conferences on Harmonisation: Guideline for dose selection for carcinogenicity studies of pharmaceuticals (1995). Fed. Reg. 60:19-28. |
| 22. | Goldsworthy, T.L., Recio, L., Brown, K, et al. (1994). Symposium overview: Transgenic animals in toxicology. Fundam. Appl. Toxicol. 22:8-19. |
| 23. | Tennant, R.W., French, J.E., Spalding, J.W. (1995). Identifying chemical carcinogens and assessing potential risk in short-term bioassays using transgenic mouse models. Environ. Health Perspect. 103:942-950. |
| 24. | Ashby, J., Tennant, R.W. (1991). Definitive relationships among chemical structure, carcinogenicity and mutagenicity for 301 chemicals tested by the U.S. NTP. Mutat. Res. 257:229-306. |
Is a New Cancer Risk Assessment Paradigm Needed?
Ronald W Hart* and Angelo Turturro
*Division of Biometry and Risk Assessment, HFT-20,
National Center for Toxicological Research, 3900 N.C.T.R.
Drive, Jefferson, AR 72079-9502; Tel. (501) 543-7232;
FAX (501) 543-7332; e-mail: rhart@nctr.fda.gov
Introduction
A new appreciation for the impact dietary intake and nutrition can exert on estimations of toxicity (1-3), has led to questions about the appropriateness of our present methods for estimating the risk of cancer in humans from chemicals shown to be positive in chronic animal bioassays. Most low dose extrapolation methodologies assume that cancer induction occurs through a non-threshold mechanism.
Dietary intake is a model non-monotonic agent. If the total intake is too low, the animal dies from protein-calorie malnutrition prior to cancer induction. As caloric intake increases above an optimum level the risk of cancer increases, consistent with elevated body weights (BW). Using dietary intake as a "model non-monotonic compound" (i.e. one which does not have a monotonic dose-response relationship) allows investigators to address a number of important issues.
Is there a threshold?
One of the issues that almost always arises when low-dose modeling is discussed is whether or not the agent of concern exhibits a threshold. A number of arguments assume that:
a) the only time to which the test subject is exposed is during the study; and b) the agent under evaluation acts independently of all other agents. These assumptions need to be reconsidered in light of new findings (4-6).
Relative to the former assumption analytical procedures have become so sensitive that many toxic agents can be found ubiquitously in the environment. For example, environmental agents created by natural processes, such as dioxin are found universally at some level of concentration (5, 7). There are multiple carcinogens and anti-carcinogens in the human diet, some of which are "introduced" by man, others which occur naturally (8). For instance, the dose of dinitropyrenes (potent mutagens) contained within an 8 oz. serving of cooked chicken is greater than that received from a full year of exposure to diesel exhaust laden air in an automotive tunnel (9).
The concept of zero exposure is, thus, untenable for many agents, and should be replaced, as has been done in an analytical context, by the concept of a level too low to measure. Thus, theoretical concerns about the efficacy of a single molecule of an agent to induce cancer in an individual have become moot. Like calories, chemical exposure appears to be unavoidable.
Relative to the latter assumption, if carcinogens and anti-carcinogens are ubiquitous then the concept that an agent can induce toxicity in isolation becomes untenable. There are no cases in vivo where there are not exposures to other agents. The significance of these other exposures will vary, from minimal to significant. Of such interactions the most significant may be with the fuel source of the body itself the common Calorie. Recently, it has been shown that modest differences in the body weight (BW) of animals on test (resulting form ad libitum feeding) result in significant differences in the tumor spectrum and time-to-tumor of these animals (1, 6, 10). Since body weight differences correlate adversely with many of the parameters important to maintenance of homeostasis, including agent disposition and pharmacokinetics, metabolism, DNA repair, DNA replication, gene expression, induced cellular proliferation, apoptosis, immunocompetency (etc), it is not surprising that a reduction in caloric intake can significantly decrease both acute and chronic toxicity. Even modest changes in BW (10-15%) can significantly alter agent toxicity (12).
The consequences of these discoveries include the necessity to evaluate the threshold of an agent in the context of the organism's environment. For example, if a threshold for the toxic action of a chemical is being evaluated, frank recognition of the effect of the chemical on other exposures should also be evaluated. This is especially critical for changes in BW, since one of the major criteria in defining MTD (the highest dose used in a toxicity test) is a significant decrement in BW gain. There are some post-hoc techniques to accommodate these effects, for instance one could use results from studies in order to attempt to mathematically adjust the results observed in the study under review. However, since the modulation of caloric intake/BW has such a plethora of effects and these effects may be highly complex, a better technique might be the use of target default growth curves. Dietary control, the control of BW growth curves by the manipulation of dietary intake, has been suggested as a means to address the variabilities induced by diet during bioassay studies (13, 14). Suggestions for target growth curves have also been made.
The type and amount of information needed to determine individual thresholds (history of diet, history of BW, individual response to exposures in the environment, etc.) is almost impossible to obtain unless controlled for at the outset of a study. However, even if such information was generally available, the possibility of antagonist and synergistic effects between these factors cannot be overlooked. The interaction of caloric intake with both normal body processes and chemical toxicity appears to cross sex, strain, and species (1, 15, 17). Such similarities in rodent and man to toxicity modulation by dietary intake is one of the strongest arguments for the appropriateness of the extrapolation of study results from animals to man and suggest that similar considerations are important in asking whether thresholds for toxic effects occur in humans.
Thus, it appears that the concept of threshold can only be realistically defined for populations under specified conditions, and should be framed in terms of the number of people in an exposed population that become either more likely to express the consequence of exposure to a toxic agent, such as cancer or the early onset of cancer, as a result of exposure to an agent. The United States, almost by default, appears to have accepted the concept that an increase in population cancer risk of 10-5 is the threshold of concern (7). This acceptance, again by default, results in a de facto threshold level of an agent. It has recently been shown that use of the current quantitative low-dose extrapolation methods to derive this level of risk is equivalent to dividing the maximally tolerated dose (MTD) by 780,000 (18).
A threshold may then be viewed as that level of agent which generates measurable toxic effects in a population under some specified set of conditions (including other toxicants prevalent at the time). In extrapolation of these results to humans, the variability in the human population of interest needs to be evaluated. Estimation of the exposures of the population of interest to other toxicants should be made, as well as consideration of what toxic endpoint and safety factors should be used to define safety.
What is the Effect of Hormesis?
Another aspect in the debate on the appropriateness of the present risk assessment paradigm which should be re-evaluated in light of the findings in diet and nutrition is the role played by hormesis, i.e., where a low-dose of certain toxicants may have a salutary effect, while higher doses are toxic (5, 19). This phenomenon is not simple to evaluate. A number of mechanisms have been suggested for hormetic effects (5, 19).
The relationship of BW to survival is one of the most obvious non-monotonic relationships that is known. This relationship is schematized in Figures 1A and 1B. The negative effects of BW on survival can be thought of as consisting of two processes. The first process is malnutrition both of micronutrients, such as vitamins and minerals, and at the lower BW of macronutrients such as protein and calories. Under these conditions a low BW results in early death. The latter process has the effect of enhancing the expression of chronic diseases, which also results in increased early death. These multiple processes combine to produce a non-monotonic dose-response relationship of BW to survival. Since this relationship is non-monotonic, any factor which influences BW can appear non-monotonic. This impact combines with the genetic propensity of the strain for certain diseases and the ability of the agent to induce disease, leading to a requirement for careful analysis of changes in BW in a chronic toxicity test for proper interpretation.
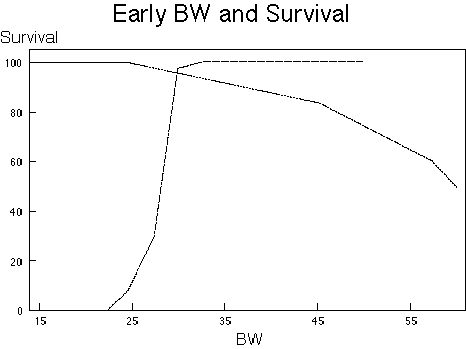
| A. | Effects of BW by Individual Process. The dotted line represents the effect of diet on survival as a result of an increase in degenerative diseases and cancer, using the effects of BW on B6C3F1 male mice as a model (11, 14). The dashed line represents the effect of protein-calorie malnutrition based on general experience at the National Center for Toxicological Research for that mouse. |
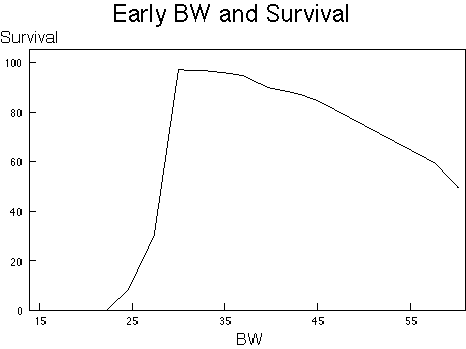
| B. | The Resultant Effect of Survival. Derived by simply adding up the two processes defined. It should be appreciated that early BW are especially important to predict survival in this mouse (20). |
| Figure 1. | Schematic View of the Effects of Body Weight (BW) on Survival in Mice. |
To illustrate how difficult it is to do a proper interpretation of this factor, a recent re-analysis of the classic work of Trotter on the hormetic effect of very low doses of radiation was undertaken (20). Exposure to low doses (0.11 rad) of radiation resulted in significantly improved survival when compared to control animals. The re-analysis noted that an effect of the low radiation dose was to inhibit the BW gain of animals early in the test, and that this inhibition, based on work in another hybrid mouse, would have resulted in an increase in survival similar to that seen in this test. Thus, the time course of the BW growth curve was important to consider. The effect of BW on survival is only one of the non-monotonic effects of BW. It has been suggested that the effects of low BW on tumor incidences, especially in group-housed animals, may also be non-monotonic (12). Since BW has been shown to effect so many factors important to toxicity, the influence of any BW changes on the many mechanisms suggested for hormesis should be considered when evaluating the impact of this phenomenon on toxicity.
Conclusion
Is a new cancer risk assessment paradigm needed? Perhaps, but prior to doing so the present risk assessment paradigm needs to be modified to incorporate new scientific findings, especially the new appreciation of the impact of dietary intake on modulation of agent toxicity.
Suggested changes in the paradigm require more information than presently used. Placing the threshold within the context of specified conditions, and the definition of rational safety factors under those conditions are not trivial exercises, and when data are not available the default procedure must be conservative enough to provide safety for the general public. Suggestions for lower or higher safety factors should be based on data, and it is anticipated that deriving data relevant to certain classes of compounds should allow refinement of the safety factors used, and also stimulate the acquisition of more information on compounds of critical concern.
This modification of the processes of risk assessment to accommodate new information was strongly recommended by past major risk assessment efforts. For instance, it was anticipated by the Office of Science and Technology Policy Cancer Risk Assessment document (21) that revisions would be necessary approximately every five years. Any discipline as important and of such practical use as risk assessment must continually incorporate new information and change. Fortunately, risk assessment appears to be flexible enough to accommodate these changes.
References
| 1) | Hart, R., Neumann, D., Robertson, R. (eds.) (1995) Dietary Restriction: Implications for the Design and Interpretation of Toxicity and Carcinogenicity Studies. ILSI Press, Washington, D.C. |
| 2) | Seilkop, S. (1995) The effect of body weight on tumor incidence and carcinogenicity testing in B6C3F1 mice and F-344 rats. Fund. Appl. Toxicol. 24: 247-258. |
| 3) | Hart, R., Turturro, A. (eds.) (1993) The Impact of Dietary Restriction on Genetic Stability. Mutat. Res. (special edition) 295 # 4-6. |
| 4) | National Research Council. (1993) Issues in Risk Assessment. National Academy Press, Washington, D.C. |
| 5) | Calabrese, E.J. (eds.) (1991) Multiple Chemical Interactions. Lewis Publishers, Inc., Chelsea, Michigan. |
| 6) | Hart, R.W., Leakey, J., Duffy, P.H., et.al. (1996) The effect of dietary restriction on drug testing toxicity. Exp. Toxic. Pathol. 48: 121. |
| 7) | Graham, J.D. (ed.) (1991) Harnessing Science for Environmental Regulation. Praeger, New York City, N.Y. |
| 8) | Ames, B.N., Gold, L.S. (1990) Chemical carcinogenesis: Too many rodent carcinogens. Proc. Natl. Acad. Sci. (USA) 87, 7772-7776. |
| 9) | Hart, R.W., Fu, P., Turturro, A. (1988) Nitro-polycyclic aromatic hydrocarbons: Structural features, genotoxicity and risk evaluation. In: Politzer, P., and Roberts, L. (eds.) Chemical Carcinogens - Activation Mechanisms, Structure and Electronic Factors and Reactivities. Elsevier Science Publisher, New York, pp 264-290. |
| 10) | Hart, R., Keenan, K., Turturro, A., et.al. (1995) Caloric restriction and toxicity. Fund.Appl. Tox. 25: 184-195. |
| 11) | Turturro, A., Hart, R.W. (1992) Dietary alterations in the rate of cancer and ageing. Exper. Geron. 27: 583-589. |
| 12) | Kari, F., Abou, K. (1995) The effects of dietary restriction on the sensitivity of the bioassay. In: Hart, R., Neumann, D., et.al. (eds.) Dietary Restriction: Implications for the Design and Interpretation of Toxicity and Carcinogenicity Studies. ILSI Press, Washington, D.C., pp. 63-78. |
| 13) | Hart, R.W., Turturro, A. (1995) Dietary restriction: An update. In: Hart, R.W., Neumann, D., et.al. (eds.) Dietary Restriction: Implications for the Design and Interpretation of Toxicity and Carcinogenicity Studies, ILSI Press, Washington, D.C., pp. 1-12. |
| 14) | Turturro, A., Duffy, P., Hart, R. (1995) The effect of caloric modulation on toxicity studies. In: Hart, R., Neumann, D., et.al. (eds.) Dietary Restriction: Implications for the Design and Interpretation of Toxicity and Carcinogenicity Studies. ILSI Press, Washington, D.C., pp. 79-98. |
| 15) | Walford, R., Harris, S., Gunion, M. (1992) The calorically restricted low-fact nutrient-dense diet in Biosphere-2 significantly lowers blood glucose, total leukocyte count, cholesterol, and blood pressure in humans. Proc. Natl. Acad. Sci. (USA) 89: 11533-11537. |
| 16) | Freni, S., Turturro, A., Hart, R. (In press) Caloric intake and anthropometric measures of growth and anabolism as indicators of risk of cancer of the breast and large bowel. Human Ecological Risk Assessment. |
| 17) | Hass, B.S., Lewis, S.M., Lipschitz, D., et.al. (In press) Dietary restriction in humans: Report on the Little Rock conference on the value, feasibility, and parameters of a proposed human study. Mech. Ag. Devel. |
| 18) | Gaylor, D.W., Gold, L.S. (1995) Quick estimate of the regulatory safe dose based on the maximum tolerated dose for rodent bioassays. Regulatory Toxicol. Pharmacol. 22: 57-63. |
| 19) | Calabrese, E. (Ed.) (1994) Biological Effects of Low Level Exposures: Dose-Response Relationships. Lewis Publishers, Chelsea, MI. |
| 20) | Turturro, A., Hart, R.W. (1994) Modulation of toxicity by diet: Implications for response at low level exposures. In: Calabrese, E. (Ed.) Biological Effects of Low Level Exposures: Dose-Response Relationships. Lewis Publishers, Chelsea, MI. |
| 21) | Interagency Staff Group. (1986) Chemical carcinogens: A review of the science and associated principles. Environ. Health Perspect. 67: 201-282. |
| |
The Need for New Risk Default Assumptions
Richard Wilson
Harvard University, Department of Physics, Cambridge, MA
02138; Tel: (617) 495-3387; e-mail: wilson@huhepl.harvard.edu
The present default assumptions used by the Environmental protection Agency and other US government agencies are 20 years old. While in principle they allow for modification when there is scientific evidence to support it, in practice change has been very difficult.
In this note I address three issues;
| (1) | Are there thresholds below which there is no risk? |
| (2) | What is the proper logical role of using animal data for prediction of human risk? |
| (3) | Is the present EPA method of managing that risk appropriate? |
Thresholds and backgrounds.
The idea that cancer incidence might be a stochastic process with the probability proportional to dose dates back at least to the work of Crowther (1924). This was followed in 1928 by the recommendation of the International Commission on Radiological Protection that a proportional relationship between radiation dose and cancer incidence be assumed for a Prudent Public Policy. Following the second world war this recommendation became of great interest, and was generally accepted. Cancers had only been observed at doses of 30 Rem (0.5 Sv) and higher and in some cohorts (such as the group of radium dial painters) there were indications of a threshold at about 20 Rems (0.2 Sv). Nonetheless the public perception arose that the linear - no threshold model has been experimentally derived and therefore true, in the regions of interest. This has in turn led to, or at least fed a technically incorrect view that radiation at low doses is uniquely dangerous. The concept of a linear no-threshold theory was being extended to chemical carcinogens in 1970, about the time the EPA was created. Both the very cautious EPA regulations and perceptions among much of the public led to a marked distinction between the handling of carcinogens and other toxic chemicals. The carcinogens are regulated to a risk level of one in a million per lifetime, pessimistically calculated, whereas if they were regulated as ordinary toxic chemicals, the dose might be merely kept to 1/10 of the No Observable Effect Level (NOEL) which is usually two orders of magnitude higher. Therefore it becomes of crucial importance to a user of a chemical to ensure that his chemical is not listed as a carcinogen. Often this is done by argument about one or two tumors in an animal study. This has led industrialists, and a number of scientists sympathetic to them, to reexamine the threshold concept.
As I consider this myself I find that I cannot separate the consideration of a threshold from the understanding of the causes of the "natural" or "background" cancers or other illnesses. The simplest and most naive way of seeing this is to imagine that we can argue that most carcinogenic substances display a threshold, and that exposures are low. Why, then is there so much cancer around? Until we can answer that question, arguments about thresholds seem hollow. A more careful and sophisticated argument appeared in a seminal paper by Crump et al. (1974) who pointed out that if a pollutant produces cancer by the same mechanism as the background cancers, there results a linear relationship between the incremental dose and the incremental impact almost independently of the biological model relating dose to response. This was used by the EPA as partial justification for their default assumption of linearity. This seminal paper has received remarkably little attention, and few people (not even the EPA) have discussed how the concept works in practice. At a minimum, the concept of incremental linearity should shift the discussion from biological thresholds (which are almost impossible to verify at the levels of interest) to a discussion of whether the pollutant and whatever causes the background cancers act by the same mechanism. This discussion need not be limited by the low statistical accuracy of epidemiological experiments or animal experiments. Nor is it a new issue. As early as 1938 Mereweather, reporting to the UK government about asbestos, mused: "Is it asbestos or the asbestosis that it causes, responsible for the lung cancers?" For asbestosis is not a natural condition, and if it is an essential step toward asbestosis caused lung cancer a threshold or non-linear dose response is plausible.
In several non-cancer medical end-points a no-threshold dose response has been suggested (Evans et al. 1982, Pease et al. 1992). Crawford and Wilson (1996) have noted that the conclusions of Crump et al. are completely general; they apply equally well to carcinogens and non-carcinogens. The one requirement is that there be a reasonably large background of the medical effect under consideration, and that the pollutant act in the same way as the background. It is evident that this might be satisfied by a large number of medical effects and pollutants.
Therefore I arrive at a belief that ANY CHANGED PARADIGM FOR REGULATION SHOULD NOT DISCUSS THRESHOLDS WITHOUT DISCUSSING BACKGROUNDS AT THE SAME TIME. Also the DISTINCTION BETWEEN REGULATORY TREATMENT OF CARCINOGENS AND NON-CARCINOGENS that only the former has a linear dose-response is probably incorrect.
(2) It is axiomatic that in consideration of new chemicals it is not permissible to carry out lifetime experiments on people. For over a century experiments on rodents have been used to provide information for toxicologists. The way in which they have been used for cancer prediction has been a matter of controversy.
In many cases tumors appeared in rodents at the same site in animal and man, or in two different species of rodent. For example exposure to vinyl chloride led to angiosarcomas in both Sprague Dawley rats and in man BUT NOT IN MICE. When tumors appeared in the same site in two species of rodent, it seemed wise to believe the likelihood that they would also appear in man at the same site. In the 1970s this was expanded when it was observed that correlations in carcinogenicity between species are much better if the site is not specified. Although the meaning of such correlations is far from clear it became regulatory policy to assume that such chemicals are carcinogenic at some site in people. But this has recently come under renewed criticism.
Starting with kidney tumors in rats, it has been pointed out that there are toxicological processes that occur in a rodent that do not occur in man. Thus, so runs the criticism, if a substance produces cancer in a rodent by such a process, it is irrelevant to the question of whether the substance causes tumors in man. I believe this is too strong a statement.
I point out that the inverse is likely. There are probably toxicological processes in man that do not exist in the species of rodent studied. For prudent public policy it is necessary to protect the public from these situations also. If I follow the simple recommendation of the critics and ignore substances where the rodent toxicological pathway does not appear in man I will fail to protect the public from these threats. However this can be resolved by a more careful understanding of the logical process, and emphasis on POTENCY rather than the binary discussion carcinogen or non carcinogen that has dominated much of the debate.
I suggest that the strong interspecies correlation of carcinogenic potency is, in some ill defined sense, a consequence of the idea that the carcinogenic potency is mostly determined by the general activity of the chemical. The toxicological pathway that can differ from species to species determines the specific site. The relationship between carcinogenic potency and acute toxicity, first noticed by Parodi et al. (1982), and amplified by Zeise et al. 1982) suggests this idea. Carcinogenic potency varies a factor of 100 million between substances, whereas the ratio of carcinogenic potency to acute toxicity (inverse of LD50) varies a factor of 100. To the extent that this is true, or something similar is true, we should NOT withdraw from regulatory attention a chemical which shows potency in one species by at a site and by a toxicological pathway that is less applicable to humans. The same chemical may well show potency at another site. But we might consider that a potency derived when the site is different is more uncertain than when the toxicological pathway is the same. There may be a smaller interspecies potency factor - but no more than a factor of 10 smaller. In some cases one might dispense with the bioassay provided that one provided similar regulation on acute toxicity (Zeise et al. 1984). This would be a way of narrowing the regulatory distinction between carcinogens and non-carcinogens in (1) above.
Crouch and myself have many times over the last 20 years suggested that the EPA should modify their procedure for estimating a potency between species by emphasizing the variability of the interspecies relationship between chemicals. These changes in the default paradigm could be made at the same time.
(3) The above paragraph is part of a general problem of how to include impacts about which we are uncertain. The present EPA procedure is to ignore the possible carcinogenic risk if the information does not exceed a certain threshold - that a chemical is categorized in a certain way. Yet once this threshold of information is exceeded, EPA attempt to present a "conservatively calculated" risk. An example of a problem that has been ignored for 10 years are the data, admittedly from an ecological study, that exposure to arsenic produces bladder and other cancers with a linear dose response relationship. If true the risks of drinking water in many western cities is orders of magnitude greater than many risks EPA regulate. EPA has been struggling to ignore these data for the last 10 years.
In the last few years there have been many calls for "scientific methods of risk assessment". When these are calls for more accurate determination of exposure (and therefore dose) they are most welcome. All too often these calls ignore the problem of how to address risks that are in some degree speculative. The risks of arsenic, of particulate air pollution and of many risks that only seem to affect old and sick people are often explicitly ignored. Many are important potential hazards and to ignore them would be very unscientific. Such calls often forget that the word "risk" does not imply a hazard that is certain, but the chance of a hazard. If there is scientific uncertainty about the existence of a hazard, there nonetheless exists a risk.
All of these point to the most important necessary change in the EPA default paradigms. Embattled industry should choose this as the primary target. Most countries of the world only regulate risks that are larger than 1 in 1,000,000 per year or 1 in 10,000 per lifetime. I maintain that the EPA cannot regulate smaller risks consistently, and any such attempt must be arbitrary and capricious, and probably illegal on that count. By unwise use of resources, such an attempt is also likely to increase risk elsewhere. The EPA should bring its de minimis level of risk closer to world practice. A proper attention to comparison of risks illustrates this most clearly. Yet even in California the "big ones get away". Proposition 65 was a proposition for informing the public - not for regulation. It would seem that it is an ideal vehicle for public education about the ubiquity of cancer and other hazards. When I was consulted many years ago about enforcement, I had one piece of advice and one only. NEVER allow an exception. My advice was ignored, and the liquor (alcohol) industry, and the restaurant (benzopyrene on charbroiled steaks) industries got their exceptions. What are left are the smaller risks. Alas the fault is ours for not standing up for common sense.
References
| 1. | Crawford M and Wilson R, (1996) "Low Dose Linearity - The Rule or the Exception?" Human and Ecological Risk Assessment 2(2) 305-330 |
| 2. | Crowther J. (1924) "Some considerations relative to the action of x-rays on tissue cells" Proc.Roy.Soc.Lond B, Biol. Sci. 96, 207-211 |
| 3. | Crump KS, Hoel DG, Langley CH, and Peto R. (1976) "Fundamental carcinogenic processes and their implications for low dose risk assessment." Cancer Res 36, 2973-2979. |
| 4. | Evans J, Ozkaynak H, and Wilson R. (1982) "The use of models in public health risk analysis." J Energy and Environ 1(1), 1-20 |
| 5. | Parodi s., Taningher M, Boero, P. and Santi, L. (1982) "Quantitative Correlations amongst Alkaline DNA Fragmentation, DNA Covalent Binding, Mutagenicity in the Ames test and Carcinogenicity for 21 Compounds" Mutation Res. 93, 1-24 |
| 6. | Zeise, L., Wilson, R, and Crouch, EAC, (1984) "Use of Acute Toxicity to Estimate Carcinogenic Risk" Risk Analysis 4, 187-189 |